We update our online model library every month and summarize the changes in the WhatsNew document. Each update makes the latest tools and improvements available to all users immediately.
In July, we continued our work to simplify tools and make them easier to apply, focusing on models that work with heat flow (Qr-Qb) data and on crystallization operations. Over the next 6 months, other application areas will receive the same review and enhancement.
We are fortunate to have Dr Wilfried Hoffmann in our team, who after nearly 29 years at Pfizer, with responsibilities and expertise ranging from thermochemistry to PAT and modeling, joined our team in 2012. Wilfried led the review of the heat flow models and the changes reflect his experience and expertise. Simple models allow rapid estimation of kinetics with very little input data and separate models translate the chemistry (by copy and paste) to larger scale conditions. Search for 'Qr' and 'exotherm' in the DCR search box to find these tools.
Decomposition reactions can be included easily alongside the synthetic chemistry reactions and safety scenarios can be explored to minimise 'accumulation' or maximize the time to maximum rate (TMR) after a cooling failure. Many of you will have seen the 2013 webinar by Bernhard Berger of Siegfried in this application area; if not, it is well worth your time to review.

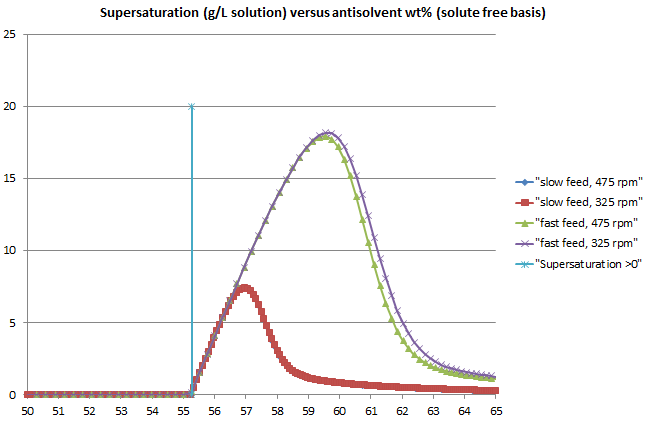
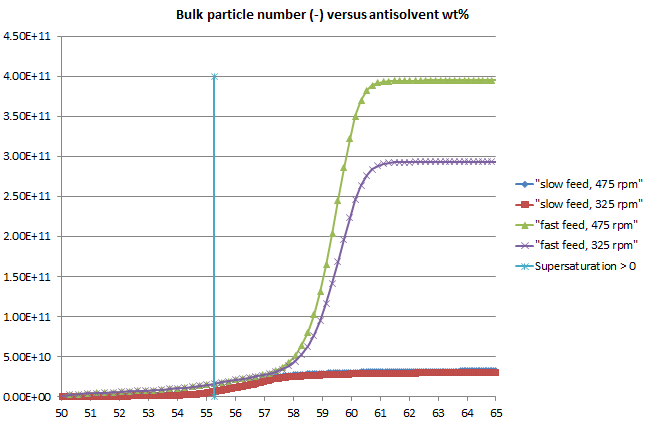
The crystallization library was also enhanced with a clearer workflow among the various tools involved and consistent, rigorous kinetics applied across all models. The previous blog post highlighted some excellent results achieved using these models, using Lasentec (CLD) data to obtain the kinetics of the true crystal size distribution (CSD), taking account of particle shape.
There will be webinars in both application areas later this year to review the improvements. See the list of current events anytime by visiting here.
In July, we continued our work to simplify tools and make them easier to apply, focusing on models that work with heat flow (Qr-Qb) data and on crystallization operations. Over the next 6 months, other application areas will receive the same review and enhancement.
We are fortunate to have Dr Wilfried Hoffmann in our team, who after nearly 29 years at Pfizer, with responsibilities and expertise ranging from thermochemistry to PAT and modeling, joined our team in 2012. Wilfried led the review of the heat flow models and the changes reflect his experience and expertise. Simple models allow rapid estimation of kinetics with very little input data and separate models translate the chemistry (by copy and paste) to larger scale conditions. Search for 'Qr' and 'exotherm' in the DCR search box to find these tools.
Decomposition reactions can be included easily alongside the synthetic chemistry reactions and safety scenarios can be explored to minimise 'accumulation' or maximize the time to maximum rate (TMR) after a cooling failure. Many of you will have seen the 2013 webinar by Bernhard Berger of Siegfried in this application area; if not, it is well worth your time to review.
The crystallization library was also enhanced with a clearer workflow among the various tools involved and consistent, rigorous kinetics applied across all models. The previous blog post highlighted some excellent results achieved using these models, using Lasentec (CLD) data to obtain the kinetics of the true crystal size distribution (CSD), taking account of particle shape.
There will be webinars in both application areas later this year to review the improvements. See the list of current events anytime by visiting here.